Online Material
Welcome to the online material for the Introduction to Research Computing on Palmetto workshop.
Slides
The slides are available in Box.
What is Palmetto
Palmetto is a high-performance computing (HPC) cluster. The cluster is made up of over a thousand computers, called nodes, connected through networks called interconnects. It has various network mounted file systems, including home and parallel scratch storage systems. A scheduler coordinates the allocation of compute resources across the cluster.
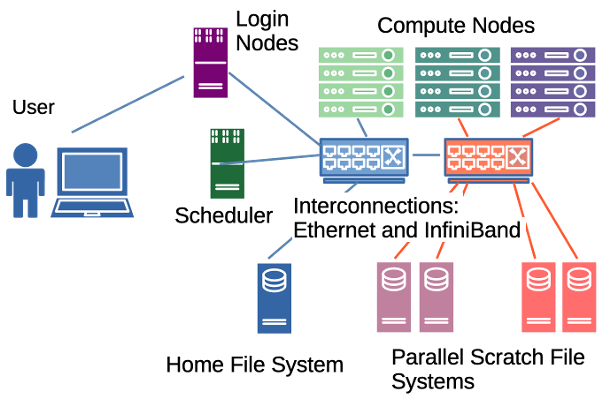
When you connect to Palmetto, you do not connect directly to the Scheduler nodes. Instead we connect to the Login nodes and from there have access to the file systems and scheduler.
Accessing Palmetto
Connect to Palmetto. Follow the steps laid out in the documentation to connect via SSH to Palmetto. On Windows, you will need to install MobaXterm.
Check the hostname. Run the following command:
hostname
What do you see?
Sample Output
You should see something like login001.palmetto.clemson.edu. You may be on a
different login node. We currently have 3 running (login001, login002,
login003). All of these possibilities indicate you are on a login node.
Resources
Each compute node has a series of resources that can be requested by jobs. There are two types:
- Consumable: only one job can use it at a time
- CPU Cores
- Memory
- GPUs
- Features: these resources are features of the node, one job requesting it
will not prevent others
- Interconnect type
- CPU Model
- GPU Model
Check resources available on by running:
cat /etc/hardware-table
This file lists information about all the compute nodes installed in Palmetto. Each row in this file represents a phase. Each phase includes a group of compute nodes that were all installed at the same time.
How many nodes are in phase 1a?
There are 117 nodes in phase 1a. Each row represents a phase.

How many nodes have P100 GPUs?
There are 60 nodes with P100 GPUs. 40 in phase 16 and 20 in phase 17. Each node has 2 GPUS, so there are a total of 120 P100 GPUs in the cluster.
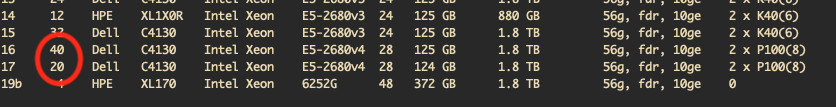
How many cores are in each node of phase 29?
Each node in phase 29 has 64 cores.
Let's check how much of the hardware is in use. To do that run
whatsfree
The output from whatsfree is similar to the format of
cat /etc/hardware-table. You get the same one row per phase information. In
addition to the "TOTAL" count, however, you also get a "FREE" count -- the
number of nodes in that phase that are completely free.
This command lets us see how busy the cluster is.
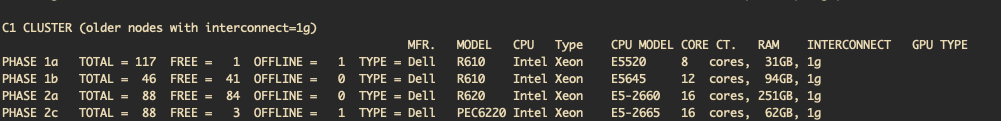
How many phase 1a nodes are free?
It changes minute to minute!
When this workshop was created, only 1 phase 1a node was free:
Storage
We have detailed documentation that covers much more that what we'll look at in this workshop.
Check to see what directory you are in. Run:
pwd
You should see /home/<username>.
If you see a different directory...
Go back home by running:
cd $HOME
Check the quota of your home. Run:
checkquota
Since most of you are new users of Palmetto, you should be using very little storage at the moment. Your max quota should read 100G.
Check the contents of your home directory. Run:
ls
This may print nothing -- that is ok! You may not have any files in your home directory yet.
File Transfer
We offer many data transfer options, depending on the file you would like to transfer.
Let us test file transfer using SFTP. First, download the following two files:
Next, upload them to palmetto.
-
On a Mac: open a (new) terminal, then run (make sure to replace your username):
cd ~/Downloads
scp compute_pi.R parallel_compute_pi.R username@hpcdtn01.rcd.clemson.edu:/home/usernameNoteThis terminal should be separate from the terminal used to run ssh. This command must be run from your local computer, it should not be run on the login node.
Complete documentation is available in our Palmetto documentation.
-
On Windows: use the built-in file browser on the left. Use the green arrow to upload the
compute_pi.Randparallel_compute_pi.Rfile you downloaded.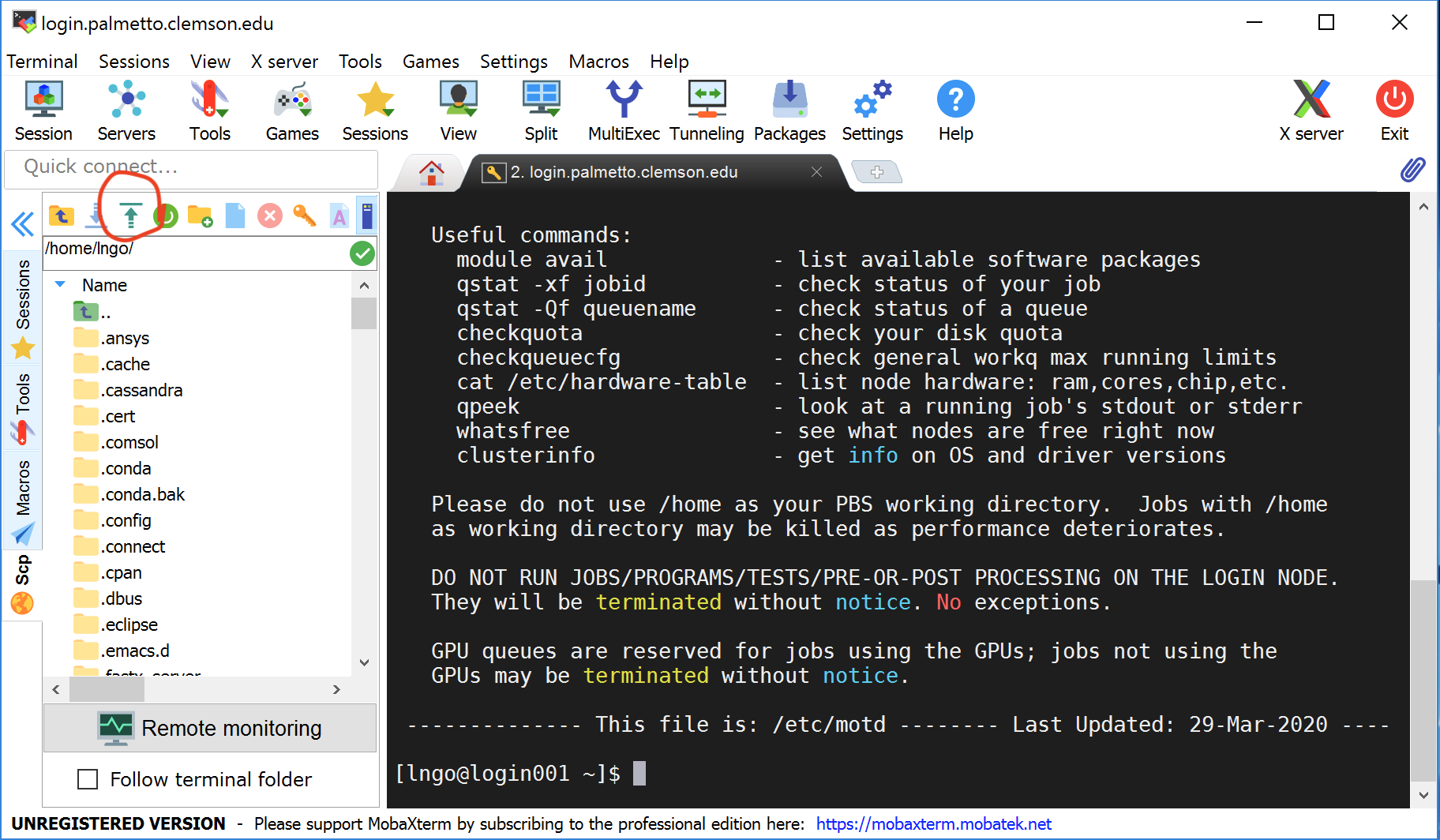
Complete documentation is available in our Palmetto documentation.
Finally, confirm the files are present by running
ls
in the terminal connected to the login node. You should now see compute_pi.R
and parallel_compute_pi.R.
Scheduling Jobs
To start a job on Palmetto, we use qsub to request resources from the
scheduler.
Let's request one compute node. Please type the following (or paste from the website into your SSH terminal):
qsub -I -l select=1:ncpus=1:mem=1gb,walltime=1:00:00
It is very important not to make typos, use spaces and upper/lowercases exactly
as shown, and use the proper punctuation (note the : between ncpus and
mem, and the , before walltime). If you make a mistake, nothing wrong will
happen, but the scheduler won't understand your request.
Here are all the parts:
qsubmeans that we are asking the scheduler to grant us access to a compute node;-Imeans it's an interactive job (we'll talk about it in a bit);-lis the list of resource requirements we are asking for;select=1means we are asking for one compute node;ncpus=1means that we only need 1 core on the node (since all Palmetto compute nodes have at least 2 CPUs, we might share the compute node with other users, but it's OK because users who use the same node do not interfere with each other);mem=1gbmeans that we are asking for 1 Gb of RAM; again, memory is specific to the user, and not shared between different users who use the same node);- finally,
walltime=1:00:00means that we are asking to use the node for 1 hour; after one hour we will be logged off the compute node if we haven't already disconnected.
This is not every resource specification possible. Be sure the check the Palmetto documentation for details on the resource limit specification.
When the job starts, you see some information from qsub, including the job
ID. For example:
qsub (Warning): Interactive jobs will be treated as not rerunnable
qsub: waiting for job 1332146.pbs02 to start
qsub: job 1332146.pbs02 ready
This indicates the job ID is 1332146.pbs02. These job IDs are important to report if you ever need help with a job. They allow research facilitators quickly lookup information about the job.
Checkout the node name. It is available in your prompt, our you can run:
hostname
Look at details of the node your requested. Run:
pbsnodes <node-name>
This command shows us all the other jobs running on this node as well as all the hardware available (the total number of cores and memory). As a reminder: we only have access to what we request -- we don't have access to everything in the node.
Software on Palmetto
We have a lot of software installed on Palmetto, but most of it is organized into modules, which need to be loaded. To see which modules are available on Palmetto, please type
module avail
Hit SPACE several times to get to the end of the module list. This is a very
long list, and you can see that there is a lot of software installed for you. If
you want to see which versions of MATLAB are installed, you can type
module avail matlab
[dndawso@node0033 ~]$ module avail matlab
------------------------------------------------- /software/AltModFiles --------------------------------------------------
matlab/MUSC2018b matlab/2021a matlab/2021b matlab/2022a (D)
Where:
D: Default Module
If the avail list is too long consider trying:
"module --default avail" or "ml -d av" to just list the default modules.
"module overview" or "ml ov" to display the number of modules for each name.
Use "module spider" to find all possible modules and extensions.
Use "module keyword key1 key2 ..." to search for all possible modules matching any of the "keys".
Let's use R. To load the module, you will need to specify its full name.To see which versions of R are available, type
module avail r
This will give you a list of all modules which have the letter "r" in them
(module avail is not very sophisticated). Let's see what happens when you load
the R 4.1.3 module:
module load r/4.2.0-gcc/9.5.0
module list
Currently Loaded Modules:
1) tcl/8.6.12-gcc/9.5.0 4) openjdk/11.0.15_10-gcc/9.5.0 7) glib/2.72.1-gcc/9.5.0
2) sqlite/3.38.5-gcc/9.5.0 5) libxml2/2.9.13-gcc/9.5.0 8) cairo/1.16.0-gcc/9.5.0
3) openssl/1.1.1o-gcc/9.5.0 6) libpng/1.6.37-gcc/9.5.0 9) r/4.1.3-gcc/9.5.0
R depends on other software to run, so we have configured the R module in a way that when you load it, it automatically loads other modules that it depends on.
Computing Pi
A simple way to compute Pi is to randomly pick points in a unit for the unit square using a uniform distribution. We then detect if a point is within the quarter of the unit circle in that exists in the quadrant. To compute an estimate for Pi, we can divide the number of points inside the circle by the total number of points (which gives us an estimate for the area inside this quarter circle) and multiply by 4.
The two scripts we uploaded in the File Transfer section perform this estimate. Let's run the first one. Since the R module should be loaded, we can run:
Rscript compute_pi.R
We should see the following message:
Killed
You may even see a message like this and have the whole job die (and be kicked back to the login node):
=>> PBS: job killed: mem 1048584kb exceeded limit 1048576kb
What's happened here is that our application requested too much memory. When you exceed the memory you requested (1gb here), PBS will kill your job.
If you PBS did not kill your job, end it by typing exit. Make sure you are
back on the login node. Let's start a new interactive job with 4gb of memory:
qsub -I -l select=1:ncpus=1:mem=4gb,walltime=1:00:00
Now, let's run the compute script:
Rscript compute_pi.R
We get the following output:
-bash: Rscript: command not found
This is expected. Every time you start a new job, you get a new clean environment. Load R, then start the script. Note, we run the script now with the time command so we can see how long it takes.
module load r/4.2.0-gcc/9.5.0
time Rscript compute_pi.R
The script will take about 30-40 seconds to finish:
Estimated Pi: 3.143039
real 0m36.148s
user 0m35.212s
sys 0m0.639s
This is pretty slow. On my MacBook, I ran this same code and it took only 10 seconds. Let's see if we can speed this up.
Speeding Things Up?
Palmetto's power does not come from exceptionally fast processors. In particular, the 1g nodes are quite old and the "single core performance" is pretty low.
The real power of Palmetto comes from the ability to span across many cores (and even multiple nodes).
Let's exit out of our previous 1 core job (run exit to leave an interactive
job) and start an 8 core job:
qsub -I -l select=1:ncpus=8:mem=4gb,walltime=1:00:00
Let's time that script again:
module load r/4.2.0-gcc/9.5.0
time Rscript compute_pi.R
There should be no speed up. The reason is that R only runs a single thread (by default). A thread can only run on one core at a time so adding more cores does nothing.
Monitoring Jobs
A handy way to monitor a job's resource utilization is
jobperf. This tool runs
on the login node and we pass the job ID as an argument.
First get another SSH session so that you still have your job running, but also have a connection to the login node.
Once you have ssh session connected to the login node, run (replace username with your username):
qstat -u username
This show you all your currently running jobs. The command qstat
has many useful options
that allow you to inspect your jobs (both running and complete).
For example, I see the following
pbs02:
Req'd Req'd Elap
Job ID Username Queue Jobname SessID NDS TSK Memory Time S Time
--------------- -------- -------- ---------- ------ --- --- ------ ----- - -----
1332258.pbs02 dndawso c1_sing* STDIN 18931* 1 8 4gb 01:00 R 00:08
I see that my 8 core job has job ID 1332258.pbs02. We can now monitor it's
status with jobperf. We want to continually monitor the job while we run, so
we'll pass the -w option. This will cause jobperf to periodically poll the
job's nodes to see resource usage. I would run the following:
jobperf -w 1332258.pbs02
While the jobperf command is running on the login node, I run the following on
the compute node:
time Rscript compute_pi.R
I can see that it is using just 1 core as we theorized:
CPU Cores Memory
Node Requested Usage Requested Current Use Max Used
node0033 8 0.99 (12.43 %) 4.00 GiB 2.07 GiB 2.08 GiB (51.91 %)
To speed this up, we can use the parallel package for R. This package spins up
a series of threads and allows you to do spawn independent tasks on each thread.
This problem is well suited for parallelism since each point can be generated
and analyzed independently. The parallel_compute_pi.R script implements this.
It takes a command line argument for how many threads we should spin up. Let's
start with 4:
time Rscript parallel_compute_pi.R 4
Output:
[dndawso@node0033 ~]$ time Rscript parallel_compute_pi.R 4
Using 4 cores with 1250000 point per core ( 5e+06 points total)
Estimated Pi: 3.141347
real 0m10.669s
user 0m0.284s
sys 0m0.151s
And in our jobperf window we see it was using about 4 cores now:
CPU Cores Memory
Node Requested Usage Requested Current Use Max Used
node0033 8 3.96 (49.48 %) 4.00 GiB 512.98 MiB 2.08 GiB (51.91 %)
If we run at 8 threads:
time Rscript parallel_compute_pi.R 8
Output:
[dndawso@node0033 ~]$ time Rscript parallel_compute_pi.R 8
Using 8 cores with 625000 point per core ( 5e+06 points total)
Estimated Pi: 3.141895
real 0m6.052s
user 0m0.314s
sys 0m0.128s
And in our jobperf window we see it was using about 8 cores now:
CPU Cores Memory
Node Requested Usage Requested Current Use Max Used
node0033 8 7.86 (98.27 %) 4.00 GiB 952.65 MiB 2.08 GiB (51.91 %)
Although R has some very nice packages that enable you to split your work across multiple threads to take advantage of multiple cores, it does require code change -- it does not happen magically. If you are curious, you can take a look at the code changes needed to convert compute_pi.R to parallel_compute_pi.R by running:
git diff compute_pi.R parallel_compute_pi.R --ignore-space-change
This workload was fairly easy to parallelize. Each thread could independently generate random points and test them. Other workloads will have to be parallelized differently and some may not parallelize at all. Feel free to discuss your problem with us in office hours.
This is a pretty significant speed up. Lets go even higher, let's use 24 cores!
[dndawso@node0193 ~]$ time Rscript parallel_compute_pi.R 24
Using 24 cores with 208333 point per core ( 4999992 points total)
Estimated Pi: 3.142628
real 0m8.690s
user 0m0.336s
sys 0m0.133s
We still only have access to 8 cores. Once we use more threads than cores, the threads will start to compete causing your code to get slower.
What happens if we were to request resources from more than one node?
To test this, let's run 2 nodes, 4 cores each. Here we allocate 2
chunks and use the
place option to force each chunk to be allocated on separate nodes.
qsub -I -l select=2:ncpus=4:mem=4gb,walltime=1:00:00,place=scatter
Then, we start and run the parallel version with 8 cores (since we have 8 cores total):
[dndawso@node0300 ~]$ module load r/4.2.0-gcc/9.5.0
[dndawso@node0300 ~]$ time Rscript parallel_compute_pi.R 8
Using 8 cores with 625000 point per core ( 5e+06 points total)
Estimated Pi: 3.140942
real 0m13.176s
user 0m0.322s
sys 0m0.236s
jobperf output:
CPU Cores Memory
Node Requested Usage Requested Current Use Max Used
node0300 4 3.95 (98.86 %) 4.00 GiB 953.36 MiB 953.36 MiB (23.28 %)
node0033 4 0.00 (0.00 %) 4.00 GiB 0 B 0 B (0.00 %)
Even though we are using the parallel package, it doesn't (by default) magically span threads across nodes. For that we typically use something like MPI, Spark, or Dask.
What happens if we were to request HDR nodes?
HDR nodes are our newest nodes and have better performance per core. We should see a speed up by requesting them. Let's request 4 cores
qsub -I -l select=1:ncpus=4:mem=4gb:interconnect=hdr,walltime=1:00:00
Then, we start and run the parallel version with 4 threads:
[dndawso@node0160 ~]$ module load r/4.2.0-gcc/9.5.0
[dndawso@node0160 ~]$ time Rscript parallel_compute_pi.R 4
Using 4 cores with 1250000 point per core ( 5e+06 points total)
Estimated Pi: 3.141929
real 0m5.733s
user 0m0.174s
sys 0m0.113s
Compared to the phase 1a node completing the job in 10.7 seconds with 4 threads, this HDR node completed it in 5.7 seconds, a pretty significant speedup.
There are some reasons to consider not using HDR nodes, however.
- These nodes tend to be in higher demand, increasing the chance you will wait in a queue.
- Some of these nodes are owned by faculty members and their priority jobs may preempt your job.
- Since they are in higher demand, we monitor them more for violations of our acceptable use guidelines. Do not request a large amount of HDR resources and let them sit idle.
Can we improve the code?
Those of you who are proficient in R may have noticed our implementation was quite inefficient. The current code samples each point and tests each point independently. R is not particularly efficient with this. It is much faster to let R operate on vectors and matrices. We can vectorize our code in parallel_compute_pi.R by changing this loop:
inside_circle <- 0
for (i in 1:(num_points / num_cores)) {
x <- runif(1, 0, 1) # Generate random x-coordinate
y <- runif(1, 0, 1) # Generate random y-coordinate
# Check if the point is inside the quarter-circle
if (x^2 + y^2 <= 1) {
inside_circle <- inside_circle + 1
}
}
return(inside_circle)
Into this vectorized code:
num_points_thread <- num_points / num_cores
x <- runif(num_points_thread, 0, 1)
y <- runif(num_points_thread, 0, 1)
return(sum(x^2 + y^2 <= 1))
Now when we run this code again with 4 cores on 1g nodes, we see a massive improvement:
[dndawso@node0241 ~]$ time Rscript parallel_compute_pi2.R 4
Using 4 cores with 1250000 point per core ( 5e+06 points total)
Estimated Pi: 3.140308
real 0m1.160s
user 0m0.292s
sys 0m0.129s
This is a bigger improvement than we saw moving from 1g to our HDR nodes. Oftentimes, it is our own code that contains inefficiencies, and the more you learn about your language of choice, the better you can write efficient code.
Open OnDemand
We have an interface to Palmetto that works through a Web browser. This interface is called "Open On Demand", or OpenOD for short. To access it, go to
https://openod.palmetto.clemson.edu
You will need to login with your Clemson username and password, and perform a DUO check.

File Browsing
One convenient feature of Open OnDemand is a file browser. In the top left corner, you will see the "Files" button, which will take you to your home directory or to scratch directory. Click it and explore the files in your file system. You can use this feature to view, edit, and delete files. It is also a convenient way to upload and download the files. You can go to any folder that you have access to by clicking "Go to".
Shell
You can also use the web interface to run a terminal on Palmetto. This way,
OpenOD becomes an alternative to MobaXTerm or to the Mac Terminal application.
To start the terminal, click on Clusters, then Palmetto Shell Access:

Enter your account password and do the two-factor identification. This will bring you to the login node of Palmetto:

From here, you can run scripts on the login node (such as checkquota,
checkqueucfg, whatsfree), and request compute nodes with qsub.
There is currently an idle timeout that happens when you use the shell access feature of Open OnDemand. If you are inactive for a period of time, the shell will need to be restarted.
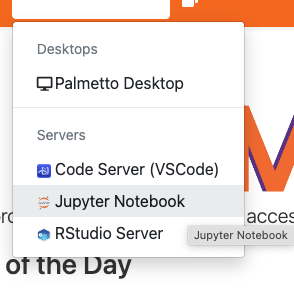
Jupyter Notebook
You can use OpenOD to run certain applications like Jupyter and Tensorflow notebooks, R Studio, and Matlab. Let's run Jupyter. From "Interactive apps", please select "Jupyter Notebook":
Please fill out the request as shown on this picture:

Cluster: Palmetto.Queue: Leave as default.Environment: Leave as Standard Jupyter Notebook.Anaconda Version: Leave as default.List of modules...: Leave blank.Conda Environment: Leave blank.CPU cores: Select 2.Memory: Select 4 GB.GPUs: Select 'None'.Interconnect: Leave as default.Walltime: Select 1 hour.Absolute path: Leave blank.
This is basically a graphical interface to qsub. You are asking for 1 compute
node, 2 CPUs, 4 GB of memory, no GPU, any interconnect, a c1 node), for the
walltime duration of 30 minutes. Once you are done entering this information,
please click the blue "Launch" button at the bottom. It will bring out a new
screen:

This means your request is being processed. Once the compute node is ready, you will see a blue button under your request saying "Connect to Jupyter":

Click on it, and it will start Jupyter.
We won't go further into Jupyter notebooks at this workshop. To exit the interactive app, you can close the browser tab, but that won't stop the underlying job. Those resources will still be locked. To stop the job, select the "my interactive sessions" icon at the top:

You should then press the Delete button on the session you are done with. This releases the resources back to the cluster so that others can use them.

Batch Job Creation
Interactive jobs are great if you need to do something quick, or perhaps visualize some data. If you have some code which runs for several hours, interactive jobs are not a great idea. Please keep in mind that an interactive job gets killed if you close the SSH connection. So for example, you connect to Palmetto from your laptop, start an interactive job, but then your laptop runs out of battery power and you can’t find your charger. SSH client quits, and your interactive job is killed.
There are ways to overcome this issue needing a persistent SSH connection (e.g. using tmux or interactive apps in On Demand), but these solutions are not ideal -- they still require you to either baby sit your job, or possibly waste lots of resources since they will not stop automatically when the computation is complete.
If you have some truly serious, multi-hour computation project (and that’s what Palmetto is really good for), a better idea is to run it on the background. This is called a batch job. You submit it in a fashion which is conceptually similar to an interactive job, but then it runs on the compute node on the background until it’s over. If it needs to take two days, it takes two days. You can quit the SSH client or close your laptop, it won’t affect the batch job.
For our example, we'll get our parallel_compute_pi.R script to run in a batch job. A batch script is typically not much more than the series of bash commands you would have run on the command line. So a great way to craft your batch job is to think about all the command your run in an interactive job. For our script we'll need to:
- Change to the appropriate directory
- Load the appropriate R module
- Run
Rscripton our script
In terms of commands, that looks like:
cd $HOME
module load r/4.2.0-gcc/9.5.0
time Rscript parallel_compute_pi.R 4
If you start an interactive job and copy and paste those lines one at a time into the terminal, you'll accomplish running the script you wanted. That is how you should think of batch scripts working.
We've almost got our script. We just have to add some header lines, making our script:
#PBS -l select=1:ncpus=4:mem=4gb,walltime=00:10:00
#PBS -N compute-pi
#PBS -j oe
#PBS -m ae
cd $HOME
module load r/4.2.0-gcc/9.5.0
time Rscript parallel_compute_pi.R 4
Each header line there is an option that could have been passed to qsub:
-l select=...: this line provides the resource requests (same as in interactive jobs)-N: allows you to name your jobs-j oe: joins the output and error messages into a single file-m ae: send an email when your jobs aborts or ends.
qsub optionsNow we have to save this as a file on palmetto. For the purposes of this workshop, we'll demonstrate the use of Open On Demand's file editor. You could instead use vim/nano over SSH or VS Code Server in Open On Demand.
In Open OnDemand, select Files->Home Directory.

Select the New File button at the top of the screen:

Enter compute_pi.pbs as the filename and press OK:

Find compute_pi.pbs and select Edit from the menu next to the file.

Paste in the contents of the batch script we developed above and select save.

Within a terminal connection to Palmetto, start the batch job:
qsub compute_pi.pbs
Note that to start the batch job, all you have to do is pass the batch script as
an argument to qsub. PBS will handle reading all the relevant options from the
file itself! qsub completes immediately, returning you to the login node. You
are never connected to a compute node as you are with an interactive job.
We can check to see that the job is running with qstat:
[dndawso@login001 ~]$ qstat -u dndawso
pbs02:
Req'd Req'd Elap
Job ID Username Queue Jobname SessID NDS TSK Memory Time S Time
--------------- -------- -------- ---------- ------ --- --- ------ ----- - -----
1898428.pbs02 dndawso c2_fdr_* compute-pi 16935* 1 4 4gb 00:10 R 00:00
Once the job is complete, it will disappear from qstat unless you pass the
-x flag:
[dndawso@login001 ~]$ qstat -xu dndawso
pbs02:
Req'd Req'd Elap
Job ID Username Queue Jobname SessID NDS TSK Memory Time S Time
--------------- -------- -------- ---------- ------ --- --- ------ ----- - -----
1898428.pbs02 dndawso c2_fdr_* compute-pi 16935* 1 4 4gb 00:10 F 00:00
The job output will be written to <job-name>.o<job-id>, so for this example
the output was written to compute-pi.o1898428:
[dndawso@login001 ~]$ cat compute-pi.o1898428
Using 4 cores with 1250000 point per core ( 5e+06 points total)
Estimated Pi: 3.14225
real 0m11.004s
user 0m0.298s
sys 0m0.140s
+------------------------------------------+
| PALMETTO CLUSTER PBS RESOURCES REQUESTED |
+------------------------------------------+
mem=4gb,walltime=00:10:00,ncpus=4
+-------------------------------------+
| PALMETTO CLUSTER PBS RESOURCES USED |
+-------------------------------------+
cput=00:00:40,mem=505324kb,walltime=00:00:13,ncpus=4,cpupercent=0,vmem=2782056kb
We see that the job ran as expected and provided us an estimated value for Pi.
PBS also prints out some handy summary statistics about the job. We can use
jobperf on the job too:
[dndawso@login001 ~]$ jobperf 1898428
Job Summary
-----------
Job ID: 1898428.pbs02
Job Name: compute-pi
Nodes: 1
Total CPU Cores: 4
Total Mem: 4.00 GiB
Total GPUs: 0
Walltime Requested: 10m0s
Status: F
Overall Job Resource Usage
--------------------------
Percent of Request
Walltime Used 13s 2.17 %
Avg CPU Cores Used 3.08 76.92 %
Memory Used 493.48 MiB 12.05 %
We see an average of 76.92% CPU usage, and only 12% memory. If I were to run this job again, I'd consider using lower memory requests.